CORRECT DIAGNOSIS:
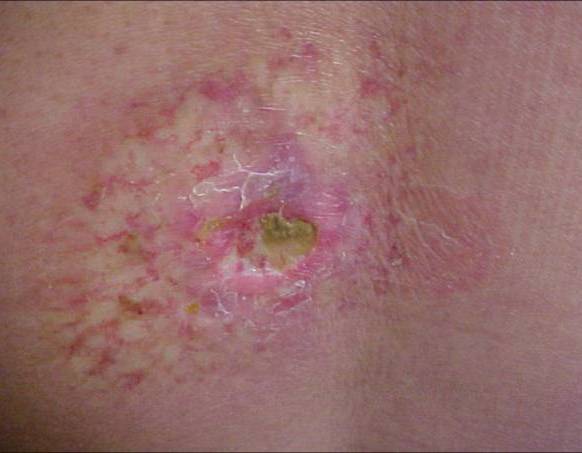
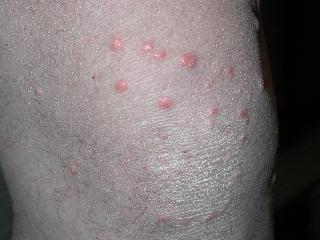
Eruptive xanthomas
DISCUSSION:
Cutaneous xanthomas are localized infiltrates of lipid-containing foamy macrophages located in the dermis and tendons. They are a marker of underlying abnormal lipoprotein metabolism, with the potential sequelae of atherosclerotic vascular disease, pancreatitis, and death. The classification of the different types of cutaneous xanthomas is summarized in Table I. Patients with eruptive xanthomas often have triglyceride levels exceeding 3000-4000 mg/dl. Genetic deficiency of lipoprotein lipase, familial deficiency of apoprotein CII, endogenous familial hypertriglyceridemia, and medicine induced elevations of triglycerides are all possible causes. Underlying medical conditions such as diabetes mellitus, hypothyroidism, renal failure, pancreatitis, obesity, and treatment with estrogens, isotretinoin, acitretin, and corticosteroids may also contribute to hypertriglyceridemia.
In North America, over 100 million people suffer from hypercholesterolemia. Despite this large number, most people with hypercholesterolemia and hypertriglyceridemia do not develop xanthomas. The exact mechanism of the formation of cutaneous xanthomas is not known. However, a possible explanation is a permeation of circulating plasma lipoproteins through dermal capillary vessels into the dermis or tendons. Subsequently, macrophages phagocytize these lipoproteins forming the classic histologic findings of foamy histiocytic cells.
Clinical features of eruptive xanthomas are characterized by yellowish papules measuring approximately 1-4mm in diameter. They are predominantly located over the extensor surfaces of the extremities, buttocks, and hands. However, the lesions can involve the trunk. An inflammatory halo may be seen in early lesions. This is thought to be due to the triglyceride component and may attribute to the tenderness and pruritus experienced by the patient. Furthermore, the Koebner’s phenomenon has been reported to occur in eruptive xanthomas.
TREATMENT:
Upon discussion of this patient with his internist, it was decided to admit our patient to the hospital for appropriate workup and initiation of lipid-lowering medication.
Control of the underlying hyperlipidemia or hyperglycemic state results in the complete resolution of the lesions. Workup should include identification of familial hyperlipoproteinemias; the most common being Frederickson type IV & V, dysfunctional apoprotein C-II, lipoprotein lipase deficiency, or impaired insulin activity.
Obesity, high caloric intake, diabetes mellitus, alcohol abuse, estrogen replacement, and retinoid therapy can exacerbate genetic defects of triglyceride metabolism and should be addressed. Complications of a missed diagnosis can result in pancreatitis or atherosclerosis.
REFERENCES:
McKee, P. H., Marsden, R. A., & Santa Cruz, D. J. (1997). Pathology of the skin with clinical correlations (pp. 7.3–7.5). Times Mirror International Publishers Limited.
Bolognia, J. L., Jorizzo, J. L., Rapini, R. P., et al. (2003). Dermatology (Vol. II, pp. 1447–1454). Elsevier.
Odom, R. B., James, W. D., & Berger, T. M. (2000). Andrews’ diseases of the skin: Clinical dermatology (9th ed., pp. 664–665).
Arndt, K. A., Leboit, P. E., Robinson, J. K., Wintroub, B. U., et al. (1996). Cutaneous medicine and surgery: An integrated program in dermatology (Vol. II, p. 1809).