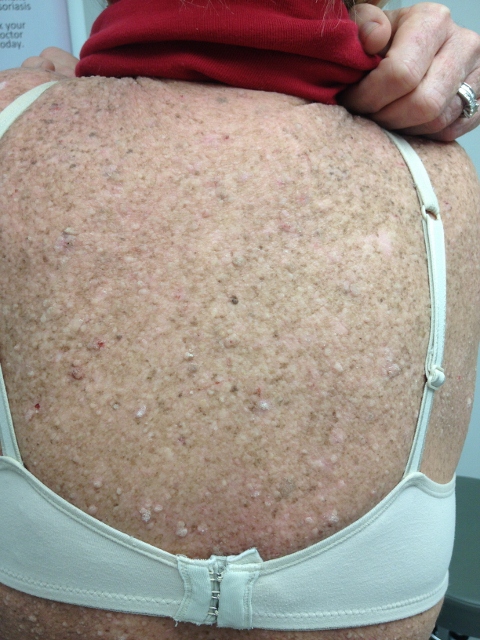
CORRECT DIAGNOSIS:
Scleromyxedema
DISCUSSION:
This case highlights the importance of scleromyxedema as a generalized papular and sclerodermoid form of lichen myxedematosus with systemic, even lethal manifestations, and distinguishes it from a localized form that does not run a disabling course. The original description of cutaneous mucinosis was described by Dubreuilh in 1906 and Reitmann in 1908. Then in 1953 Montgomery and Underwood proposed a clinical classification distinguishing four types of lichen myxedematosus: a generalized lichenoid eruption; a discrete papular form; a generalized or localized lichenoid plaque form and an urticarial form. The term scleromyxedema was first proposed in 1954 by Gottron to denote the generalized lichenoid papular eruption with sclerodermoid features. Scleromyxedema must be understood as a generalized variant of cutaneous mucin deposition with systemic, even lethal, manifestations.
Although the exact pathogenesis of scleromyxedema is unknown, various hypotheses exist. A number of immunomodulatory mechanisms have been proposed to attempt to link the monoclonal gammopathy with fibroblast proliferation however the precise relationship between skin changes and paraproteinemia remains unclear. It has been proposed that the paraprotein acts as an autoantibody and directly stimulates fibroblast proliferation and mucin deposition in the skin Harper and Rispler provided evidence against this hypothesis showing that serum of 3 patients containing paraprotein and one patient’s serum without paraprotein stimulated fibroblast DNA synthesis and proliferation in vitro. Additionally, the paraprotein did not have stimulatory effects when eluted and isolated.
Later these same results of a causal relationship between scleromyxedema patients’ serum and fibroblast proliferation could not be duplicated by another group of researchers. Instead, Yaron et al demonstrated that serum could induce a 2-fold increase in hyaluronic acid synthesis and a 13-fold increase in prostaglandin E synthesis. These findings possibly suggest a causal relationship between prostaglandin-E synthesis as a mediator that then stimulates the synthesis of hyaluronic acid. Scleromyxedema is an uncommon disease of middle age persons without sex predilection. The disease presents typically two components of the skin eruption. The papular component presents as a symmetric firm, waxy papules approximately 2 to 3 millimeters in diameter. These papules are found most commonly on the bilateral hands, arms, face, neck, upper trunk, and proximal lower extremities. The papules are typically arranged in a linear pattern. A generalized woody induration of the skin is the second component and presents similar to scleroderma. The cutaneous involvement typically spares the mucous membranes as well as the scalp. As well, telangiectasias and calcinosis are always absent.
Patients with scleromyxedema may have significant cutaneous as well as extracutaneous involvement leading to significant comorbidity associated with this disease. Hematologic disorder in scleromyxedema patients includes a paraproteinemia with rare progression to multiple myeloma. Central and peripheral nervous system involvement includes coma following a flu-like illness and paresthesias, respectively. The musculoskeletal system is occasionally affected by patients, presenting with varying degrees of proximal muscle weakness. Interestingly post-mortem examination of patients with known scleromyxedema revealed no mucin deposition in the brain and mucin deposition in the muscle in only 2 patients. Pulmonary involvement may manifest as obstructive or restrictive lung involvement. Gastrointestinal involvement presents as progressive dysphagia.
It is important to note, however, that although many patients with scleromyxedema report a wide variety of systemic symptoms, a correlation with mucin deposition on post-mortem autopsies infrequently correlates. For instance, myopathy is a common finding in patients with scleromyxedema but upon muscle biopsy mucin deposition is usually not found based on previous cases cited in the literature. Therefore something rather than mucin may contribute to the extracutaneous systems involved with this disease.
Histologically, the skin shows a diffuse deposit of mucin in the upper and mid reticular dermis, an increased collagen deposition, and marked proliferation of irregularly arranged fibroblasts. The epidermis may be either normal or thinned by the pressure of the underlying mucin and fibrosis. Hair follicles may be atrophic and a slight perivascular, superficial lymphocytic and plasmocytic infiltrate is often present. The elastic fibers are usually fragmented and decreased in number. Due to the rarity of this disease no prospective, controlled therapeutic trials are reported in the literature. Past literature involves case studies with outcomes for different therapeutic modalities implemented for particular patients. It is a disorder presenting in the skin with systemic involvement and the etiology of this systemic disease is not clearly understood. Therefore treatment of the cutaneous involvement includes topical, intralesional, and systemic steroids; topical and intralesional hyaluronidase; topical dimethyl sulfoxide; corticotropin; PUVA which may decrease skin thickness but prolonged use increases the risk of squamous cell carcinoma; Grenz ray; electron beam therapy; retinoids by possibly reducing fibroblast proliferation; plasmapheresis; dermabrasion; extracorporeal photochemotherapy. The underlying disease process; however, may be targeted by drugs used to treat other hematologic disorders such as melphalan and other chemotherapeutic agents. Due to the significant hematologic malignancies and chance of life-threatening infection, these therapies are limited to patients severely impacted by the co-morbidities associated with the disease. High-dose immune globulin has also been used after reported success in treating a neurologic disease associated with a paraprotein. Limited use with granulocyte colony-stimulating factor, cyclosporine, thalidomide, and interferon alfa has been reported in the literature. Treatment is commonly disappointing and the prognosis is overall poor.
TREATMENT:
A diagnosis of scleromyxedema was made. The patient was referred to several sub-specialists including a neurologist, hematologist/ oncologist, rheumatologist, and physical therapists. After collaboration among the sub-specialists combination therapy including an alkylating agent, melphalan, (L-Phenylalanine Mustard), and intravenous immune globulin (IVIG) was initiated. Despite the overwhelming possibility of potential side effects, these aforementioned drugs were started in light of the patient’s worsening physical condition.
Melphalan was dosed at 2 milligrams (mg) every other day for one month until she developed leukopenia and therapy was interrupted for a period of nine days. She then continued melphalan at 2 mg every other day for a total of 8 cycles. The patient’s dose was then reduced to 2 mg given only Mondays and Thursdays for 4 cycles. Finally, the melphalan was administered only on a once-weekly basis due to bone marrow suppression evidenced on repeat laboratory reports. Darbepoetin alfa support was utilized with the emergence of anemia.
Concomitantly the patient began IVIG therapy consisting of 5-day infusions given every three weeks. This particular patient received IVIG 30 grams per day for 5 consecutive days. After 12 consecutive cycles, the patient’s frequency of administration of the drug was reduced to every five weeks. The patient’s primary side effect with the administration of IVIG was nausea and mild flares of stomatitis. Toward the end of therapy, patient reported “not feeling well” all week of therapy despite premedication with 1,000 mg of oral acetaminophen.
The patient continues success with single-drug therapy managed by a hematologist/ oncologist. The patient continues successful therapy on melphalan over the past year. The success of therapy is measured by our patient’s quality of life. Her lower extremity weakness improved with increased ability to perform activities of daily living (ADLs) previously requiring assistance. During the last office visit with the patient, she now works full-time.
Serum protein electrophoresis continued to show an elevated M spike throughout the course of therapy. Muscle enzymes, including CPK, previously elevated returned to normal limits during the course of systemic chemotherapy.
REFERENCES:
Montgomery, H., & Underwood, L. J. (1953). Lichen myxedmatosus (differentiation from cutaneous myxedemas or mucoid states). Journal of Investigative Dermatology, 20, 213-236.
Dubreuilh, W. (1906). Fibromes miliares folliculaires: Sclerodermie consecutive. Annales de Dermatologie et de Syphiligraphie, 37, 569-572.
Reitmann, K. (1908). Über eine eigenartige, der sklerodermie nahestehende Affection. Archiv für Dermatologie und Syphilis, 92, 417-424.
Rongioletti, F., & Rebora, A. (2001). Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. Journal of the American Academy of Dermatology, 44(2), 245-252.
Lister, R., Jolles, S., et al. (2000). Scleromyxedema: Response to high-dose intravenous immunoglobulin. Journal of the American Academy of Dermatology, 43(2), 284-286.
Harper, R. A., & Rispler, J. (1978). Lichen myxedematosus serum stimulates human skin fibroblast proliferation. Science, 199, 545-547.
Yaron, M., Yaron, I., Yust, I., & Brenner, S. (1985). Lichen myxedematosus serum stimulates hyaluronic acid and prostaglandin E production by human fibroblasts. The Journal of Rheumatology, 12, 171-175.
Rothe, M. J., Rivas, R., Gould, E., & Kerdel, F. A. (1989). Scleromyxedema and severe myositis. International Journal of Dermatology, 28, 657-660.
Godby, A., Bergstresser, P., et al. (1998). Fatal scleromyxedema: Report of a case and review of the literature. Journal of the American Academy of Dermatology, 38(2), 285-289.
Verity, M. A., Toop, J., McAdam, L. P., & Pearson, C. M. (1978). Scleromyxedema myopathy. American Journal of Clinical Pathology, 69, 446-452.
Espinosa, A., De Miguel, E., Morales, C., Fonseca, E., & Gihon-Banos, J. (1993). Scleromyxedema associated with arthritis and myopathy: A case report. Clinical and Experimental Rheumatology, 11, 545-547.
Van Doorn, P. A., Vermeulen, M., Brand, A., Mulder, P. G. H., & Busch, H. F. M. (1991). Intravenous immunoglobulin treatment in patients with chronic inflammatory demyelinating polyneuropathy: Clinical and laboratory characteristics associated with improvement. Archives of Neurology, 48, 217-220.