CORRECT DIAGNOSIS:
Recessive Dystrophic Epidermolysis Bullosa
DISCUSSION:
Hallopeau-Siemens Recessive Dystrophic Epidermolysis Bullosa is a genetic mechano-bullous dermatosis which is often incapacitating for the 1-2 cases per million births annually. Incidence is higher in Norway and lower in Japan & Croatia. It affects both sexes equally and is characterized by autosomal recessive inheritance of COL7A1 gene mutations in which premature termination codons lead to the absence of anchoring fibrils within the Dermal Epidermal Junction (DEJ). Diagnosis is made by demonstration of separation in the DEJ below the lamina densa in the skin and extracutaneous mucosa, in addition to clinical signs of marked growth retardation, and widespread scarring. The gold standard of diagnosis is the electron microscopic findings of absent anchoring fibrils or genetic evaluation of COL7A1 mutation.
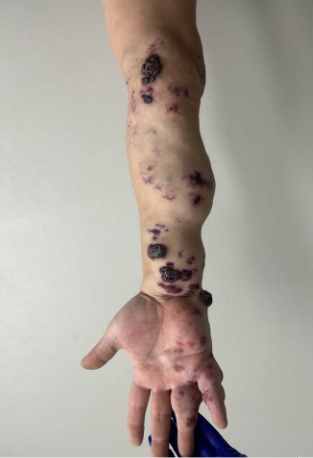
Patients have progressive, scarring disease. Blistering is generalized at birth and while life expectancy is normal, early death from overwhelming infection, metastatic scar associated squamous cell carcinoma, or malnutrition associated complications is typical. Skin findings include blisters, milia, atrophic scarring, dystrophic or absent nails, scalp abnormalities, and scar associated squamous cell carcinomas. Extracutaneous involvement includes anemia, malnutrition/ growth retardation, oral cavity soft tissue abnormalities, periodontitis, enamel hypoplasia leading to caries, gastrointestinal scarring/ stenosis, genitourinary scarring, ocular scarring, pseudosyndactyly (mitten hand deformity), and flexion contractures.
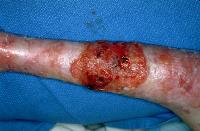
The most serious complication of RDEB is squamous cell carcinoma arising in chronic non-healing ulcers which are difficult to distinguish from the chronic ulceration, scarring, and crusting typical of RDEB. These invasive SCC’s are typically well to moderately differentiated and occur mostly on the limbs. The incidence of these SCC’s is around the ages of 15-35,1 and 50% are affected by age 20-35. These SCC’s have a high metastatic potential.
Diminished food intake and increased nutritional requirements lead to malnourishment which commonly results in mixed anemia from iron deficiency or failure to thrive. Compounding this malnutrition is the mucosal scarring that leads to strictures and webs of the esophagus, urethral and anal stenosis, and phimosis. RDEB patients have increased caloric and protein requirements, and hemodynamic and metabolic responses typical of burn patients. This disequilibrium leads to an inability to overcome infection and inadequate wound healing. Impediments to the intake of adequate nutrition include oral, pharyngeal, and gastrointestinal lesions, abnormal esophageal motility/ dysphagia, strictures, malabsorptive diarrhea, and dental problems. Nutritional deficits may lead to fatal cardiomyopathies. Fatal systemic amyloidosis has also been reported.
Ophthalmic complications affect 51% of RDEB patients including corneal scarring, lid ectropion, lid blisters, and symblepharon. These complications can be devastating for these patients and are treated with vigilant use of lubricants.
TREATMENT:
Most treatment is supportive nutrition, wound care, and prevention of trauma. Wound healing is treated by the removal of necrotic tissue or foreign body, prevention of infection by liberal usage of antibiotic ointments and nutritional support to maintain an adequate immune status. Common pathogens are Staphyloccus aureus, Streptococcus pyogenes, and Gram-negative organisms like Pseudomonas aeruginosa. Treatment regimens may include whirlpool debridement and semiocclusive, non-adherent dressings like Vaseline gauze.
Tumor surveillance is important since multiple primary squamous cell carcinomas may arise as early as 15-20 years of age. Gastrointestinal evaluation of scarring and strictures is important since nutritional requirements are high in these chronically wounded patients. Dysphagia may be managed by the use of phenytoin or oral steroid elixirs. It is also important to treat oral pharyngeal Candidiasis. Ocular lesions (as above) may include cicatricial conjunctivitis, chronic blepharitis, and corneal ulceration, erosion, and scarring. These may be treated with topical antibacterial ointments or cycloplegic agents to reduce ciliary spasm. Oral care is important to maintain nutritional intake. Aggressive preventive interventions including gentle dental hygiene are the primary treatment to prevent dental caries, which are common due to enamel hypoplasia. Water pulsation devices and very soft gentle tooth brushing are critical. Oral mucosal blisters can be treated with saline rinses and avoidance of alcohol-containing mouthwashes. Avoidance of sharp-edged food such as chips and hard candy in addition to more aggressive blenderized foods are important to maintain mucosal integrity.
Gene therapy is the only definitive treatment but is still not a part of our armamentarium. Surgical care may include the correction of esophageal strictures by balloon dilatation or colonic interposition. Aggressive active nutritional support includes starting at an early age and consideration of nasogastric or gastrostomy feeding tubes at earlier stages. Surgical correction of pseudosyndactyly followed by long term night splinting is associated with a typical recurrence in 5 years. The use of Apligraft to cover acute wounds has led to faster healing of ulcers, less pain, and better quality of life by preventing acute wounds from becoming chronic wounds. Apligraft skin is not lasting and is eventually replaced by the patient’s normal diseased skin over time. Reblistering occurs in about 20% of Apligrafted skin lesions.
REFERENCES:
McGrath, J. A., Schofield, O. M., Mayou, B. J., et al. (1992). Epidermolysis bullosa complicated by squamous cell carcinoma: Report of 10 cases. Journal of Cutaneous Pathology, 19(2), 116–123. [PMID: 1568487]
Terrill, P. J., Mayou, B. J., McKee, P. H., & Eady, R. A. (1992). The surgical management of dystrophic epidermolysis bullosa (excluding the hand). British Journal of Plastic Surgery, 45(6), 426–434. [PMID: 1511257]
Tong, L., Hodgkins, P. R., Denyer, J., et al. (1999). The eye in epidermolysis bullosa. British Journal of Ophthalmology, 83(3), 323–326. [PMID: 10227336]
Pulkkinen, L., & Uitto, J. (1999). Mutation analysis and molecular genetics of epidermolysis bullosa. Matrix Biology, 18(1), 29–42. [PMID: 10077308]
Fine, J. D., Eady, R. A., Bauer, E. A., et al. (2000). Revised classification system for inherited epidermolysis bullosa: Report of the Second International Consensus Meeting on diagnosis and classification of epidermolysis bullosa. Journal of the American Academy of Dermatology, 42(6), 1051–1066. [PMID: 10882785]
Wright, J. T., Fine, J. D., & Johnson, L. (1993). Hereditary epidermolysis bullosa: Oral manifestations and dental management. Pediatric Dentistry, 15(4), 242–248. [PMID: 8353682]
Allman, S., Haynes, L., MacKinnon, P., & Atherton, D. J. (1992). Nutrition in dystrophic epidermolysis bullosa. Pediatric Dermatology, 9(3), 231–238. [PMID: 1321222]
Terrill, P. J., Mayou, B. J., & Pemberton, J. (1992). Experience in the surgical management of the hand in dystrophic epidermolysis bullosa. British Journal of Plastic Surgery, 45(6), 435–442. [PMID: 1511258]
Falabella, A. F., Valencia, I. C., Eaglstein, W. H., & Schachner, L. A. (2000). Tissue-engineered skin (Apligraf) in the healing of patients with epidermolysis bullosa wounds. Archives of Dermatology, 136(10), 1225–1230. [PMID: 11041924]
Marinkovich, M. P., Herron, G. S., & Khavari, P. A. (1999). Hereditary epidermolysis bullosa. In Fitzpatrick’s Dermatology in General Medicine (5th ed., pp. 690–700). New York: McGraw-Hill.