CORRECT DIAGNOSIS:
Atypical Pyoderma Gangrenosum
DISCUSSION:
Pyoderma gangrenosum is a rare, inflammatory, cutaneous manifestation of systemic disease in approximately half of all affected patients. It is a diagnosis of exclusion, first described by Brunsting, Goekerman, and O’Leary in 1930. The exact etiology remains unknown, but associated conditions most commonly include inflammatory bowel diseases, such as ulcerative colitis and Crohn’s disease, polyarthritis (seropositive and seronegative), spondylitis, hematological disease (acute myelogenous leukemia; hairy cell leukemia), monoclonal gammopathies (IgA paraproteinemias/myelomas), hepatitis, SLE, Sjogren syndrome, Sweet’s syndrome, Behcet’s, and other autoimmune diseases. PG can arise before, during, or after diagnosis of any of the associated systemic conditions. Its pathophysiology is often idiopathic and poorly understood, but altered neutrophil chemotaxis is thought to play a major role in this highly inflammatory, neutrophilic dermatosis.
PG is encountered worldwide with diagnosis most commonly occurring during the 3rd to 6th decades of life. Women are more affected than men, and childhood involvement is rare (4%). Pathergy, the phenomenon in which cutaneous trauma initiates the development of the disease, occurs in approximately 30% of all cases.
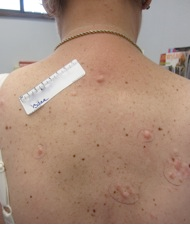
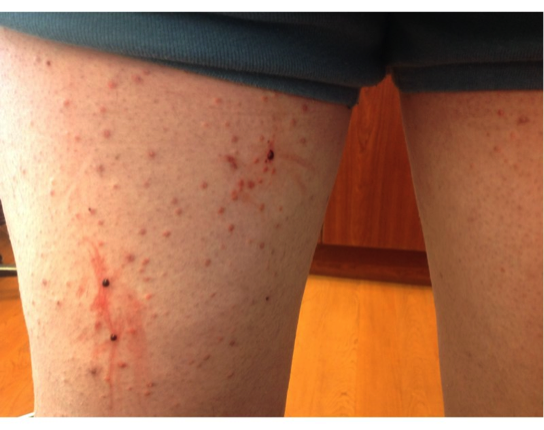
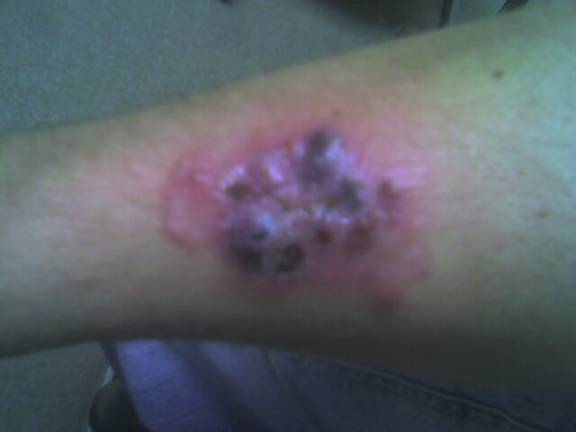
There are four recognized variants of PG. The ulcerative or classical type usually begins as a small, tender, papule or pustule on an erythematous base, which rapidly expands outward. A common misdiagnosis is that of a spider bite due to the pain and clinical presentation. The area becomes purulent and ulcerates with necrotic, undermined borders that have a gun-metal gray color. The classical type most often presents on the pretibial legs. Lesions typically heal as an atrophic, cribriform, pigmented scar. Healing may occur spontaneously or only after treatment of an underlying systemic condition, most commonly inflammatory bowel disease.
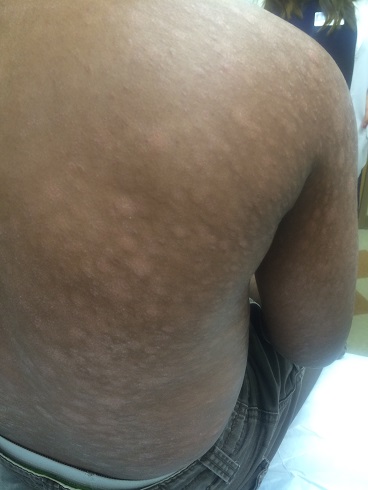
Atypical or vesiculobullous PG most often presents on the upper extremities, dorsal hands, or face. It is most often encountered in the setting of hematological malignancies such as acute myelogenous leukemia and myeloproliferative disorders such as IgA monoclonal gammopathy. It can be severely hemorrhagic and patients may have systemic symptoms of fever, myalgias, and arthralgias.
Pustular PG occurs as multiple small pustules that may or may not progress to a classical lesion of PG. Inflammatory bowel disease or Behcet’s disease may be associated with conditions.
The superficial granulomatous type of PG usually follows major trauma or surgery to the skin. There is less neutrophilic inflammation compared to the other variants and it is not aggressive. This type favors the trunk and it may be related to Wegener’s granulomatosis. Pyostomatitis vegetans is PG occurring on the oral mucous membranes. It is encountered in the setting of inflammatory bowel disease. Other sites include peristomal, labial, vulvar, scrotal, and perianal areas. With all of the above types of pyoderma gangrenosum, neutrophilic inflammation may occur extracutaneously. Neutrophilic infiltrates may affect the lungs (most common), heart, bones, eyes (peripheral ulcerative keratitis), GI tract, liver, pancreas, spleen, kidneys, lymph nodes, and central nervous system.
Differential diagnosis is numerous and includes vascular occlusive disease, vasculitis, carcinoma, infection, exogenous tissue injury, insect bite, ecthyma, sporotrichosis, Sweet’s syndrome, Churg-Strauss Syndrome, Wegener’s granulomatosis, tuberculosis gumma, syphilitic gumma, deep fungal infection, factitial disease, and others. In order to accurately diagnose PG, it is imperative to rule out other diseases via a comprehensive history, physical examination, and medication review. A biopsy should be obtained from an advancing edge of the lesion and special stains employed. Histopathological findings in PG are non-specific, but usually include massive neutrophilic inflammation, hemorrhage, and necrosis. It is necessary to culture for bacterial, viral, fungal, and mycobacterial organisms. Basic laboratory evaluation includes a CBC with differential, a comprehensive metabolic panel with liver function tests, hepatitis panel, and urinalysis. If PG is considered, obtain serum and urine protein electrophoresis, ANA, ANCA, antiphospholipid antibody, and an RPR. A CXR and GI tract studies may be recommended as well.
Treatment of PG is usually directed at the underlying systemic illness if present. In those cases in which no underlying systemic illness can be found, there are many various treatments available. In addition to topical therapies including superpotent steroids, cromolyn sodium 2%, tacrolimus, pimecrolimus, and topical and oral antibiotics, the standard treatment of choice is oral systemic corticosteroids at a dosage of 1-2mg/kg/day or pulsed IV methylprednisolone until resolution. In steroid unresponsive cases, other immunosuppressants may be used. These include cyclosporine, azathioprine, mycophenolate mofetil, cyclophosphamide, chlorambucil, tacrolimus, intravenous immunoglobulin (IVIg), thalidomide, etanercept, infliximab, adalimumab, clofazimine, dapsone, metronidazole, colchicines, methotrexate, SSKI, and nicotine. In a recent study published in the journal Gut, 69% of patients treated with infliximab at an infusion dose of 5mg/kg improved clinically whether they had associated inflammatory bowel disease or not. Additionally, local wound care is employed to prevent secondary bacterial contamination and sepsis. Hyperbaric oxygen may be used to assist in the healing of refractory cases. Surgical debridement should be avoided due to the risk of pathergy.
Recurrence and chronicity is not uncommon.
TREATMENT:
Following the biopsy, the patient was empirically treated with cephalexin (Keflex) 500mg TID and mupirocin 2% ointment to the affected area TID. He was informed that the most likely etiology related to his underlying ulcerative colitis. His primary physician was contacted and the information shared revealed a non-compliant patient with a history of pan-colitis. He was referred back to his primary care physician for evaluation and treatment of ulcerative colitis and full laboratory evaluation to exclude other potential etiologies. His primary physician prescribed azathioprine (Imuran) 50mg daily, mesalamine (Asacol) 800mg BID, and prednisone 40mg daily. Two weeks later, his follow up visit examination revealed marked improvement. The lesion had regressed to a 4.0 x 2.5cm slightly erythematous, scaly, atrophic patch with evidence of cribriform scarring. He denied tenderness or pruritus.
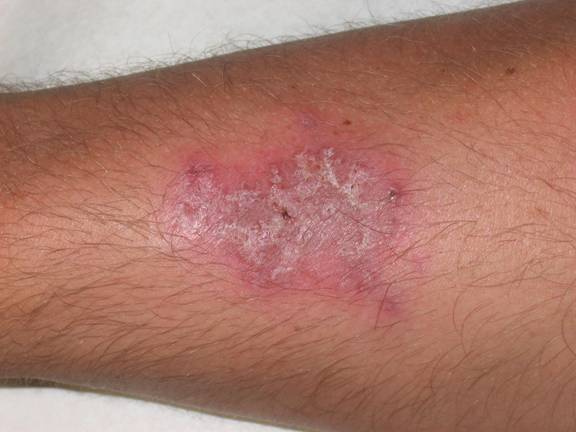
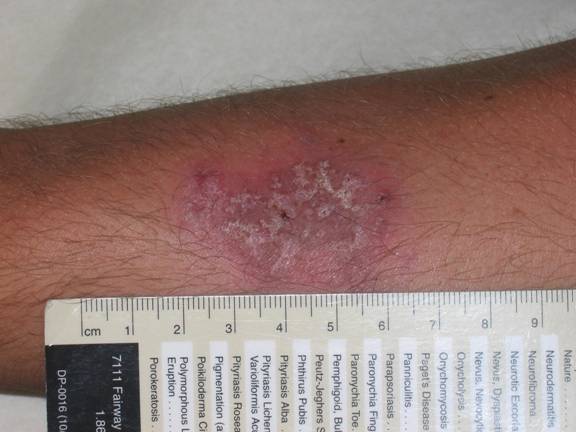
The patient in this case was diagnosed with atypical pyoderma gangrenosum based on clinical and histopathological findings, as well as the history of ulcerative colitis and non-compliance with medical treatment. The hypothesis is that his occupation as a sandblaster placed him at risk for superficial injury to unprotected skin. He reported that he often wore little protection on the job and small dust particles were always bombarding his skin. These injuries most likely resulted in pathergy of the involved extremity and initiation of the inflammatory process. A detailed discussion stressed the need for compliance with therapy and protection of his skin while working as a sandblaster. Clobetasol 0.05% (Temovate) cream was prescribed BID and he was told to follow up in 2 weeks. The patient is assumed to have resolved completely as he has not kept any subsequent appointments.
REFERENCES:
Bolognia, J. L., Jorizzo, J. L., Rapini, R. P., et al. (2003). Dermatology. Spain: Mosby.
Brooklyn, T. N., Dunnill, M. G. S., et al. (2006). Infliximab for the treatment of pyoderma gangrenosum: A randomized, double-blind, placebo-controlled trial. Gut, 55, 505–509. PMID: 16227571
Callen, J. P. (1998). Pyoderma gangrenosum. Lancet, 351, 581–585. PMID: 9472358
Callen, J. P. (1989). Pyoderma gangrenosum and related disorders. Medical Clinics of North America, 73(5), 1247–1261. PMID: 2671087
Chow, R. K. P., & Ho, V. C. (1996). Treatment of pyoderma gangrenosum. Journal of the American Academy of Dermatology, 34, 1047–1060. PMID: 8657887
Jackson, J. M., & Callen, J. P. (2006). Pyoderma gangrenosum. eMedicine. Retrieved from [eMedicine website].
Odom, J. (2000). Andrew’s Diseases of the Skin (9th ed.). Philadelphia, PA: Elsevier.
Perez, E., & Watsky, M. (2006). Pyoderma gangrenosum: A case study and review of treatment options. JAOCD, 1, 70–72. PMID: 18265067
Powell, F. G., Su, W. P. D., & Perry, H. U. (1996). Pyoderma gangrenosum: Classification and management. Journal of the American Academy of Dermatology, 34, 395–409. PMID: 8657865
Weenig, R. H., & Davis, M. D. P. (2002). Skin ulcers misdiagnosed as pyoderma gangrenosum. New England Journal of Medicine, 347(18), 1412–1418. PMID: 12393887