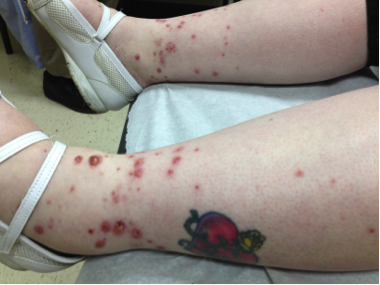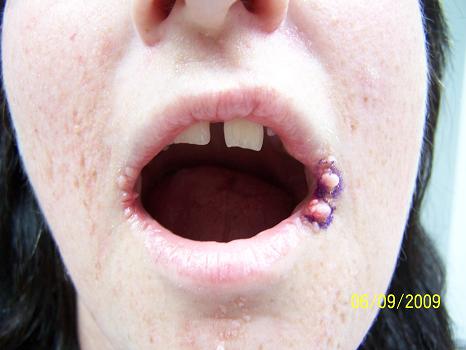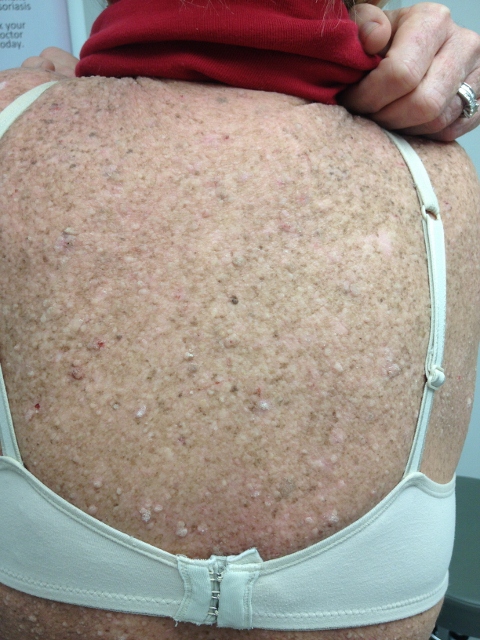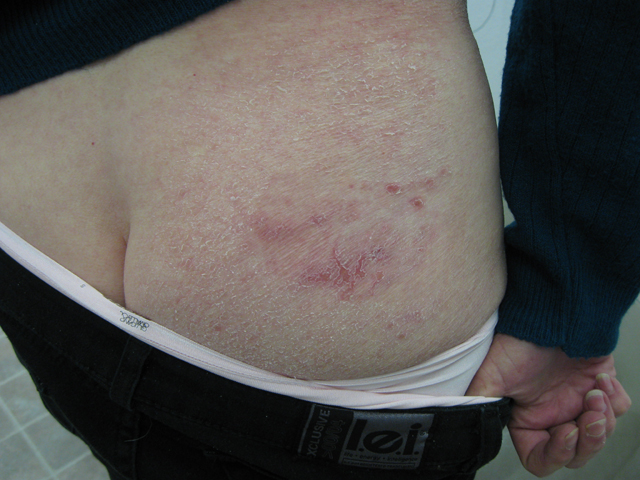
CORRECT DIAGNOSIS:
Mycosis Fungoides
DISCUSSION:
Mycosis Fungoides, despite its name, is not a fungal disease. It is the most common type of cutaneous T-cell lymphoma (CTCL) and is typically diagnosed in the 5th or 6th decade of life. Mycosis Fungoides is more prevalent in men than in woman and in African Americans versus Caucasians. The etiology remains a mystery although genetic, immunologic, and environmental factors have been implicated.
The disease course is often lingering and evolves over several clinical phases: pre-MF, patch, plaque, and tumor. Lesions are most common in the bathing suit area and can be pruritic. The pre-MF phase is commonly misdiagnosed as eczema or when biopsied as non-specific spongiotic dermatitis. It can take up to 35 years for the lesion to progress into a patch or plaque phase with the median length to diagnosis around 5 years.
Distinguishing histological features depend on the clinical stage. Early lesions demonstrate patchy bandlike lymphocytic infiltrate in the papillary dermis with coarse fibrosis. Epidermotropism is a common feature and Pautrier microabscesses filled with atypical lymphocytes containing cerebriform nuclei are characteristic of MF. Immunophenotyping often reveals CD2+, CD3+, CD4+, CD5+ and CD8- with loss of CD7. T cell receptor gene rearrangement is typically clonal.
Staging of MF, like most cancerous conditions, is done using the TNM system: Tumor (clinical phase and percent of involved skin), Nodal (lymph node involvement), Metastasis (visceral organ involvement) and in addition, also includes Blood evaluation (presence of Sezary cells). Prognosis depends on staging, with infection or systemic involvement accounting for the cause of death.
Our patient is T1, N0, M0, B0 placing her in the clinical stage IA which carries a 95% five-year survival rate.
TREATMENT:
Our patient was referred to an oncologist for a full systemic workup. She declined treatment of the lesion at this time which could include topical corticosteroids, topical chemotherapy (nitrogen mustard or carmustine), topical bexarotene and PUVA or UVB. Additional treatment options for more progressive cases include radiotherapy, systemic multiagent chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone), interferons, systemic retinoids, and other biologic response modifiers. However, it is important to note that treatment has not been shown to prolong life.
REFERENCES:
Arnold, Odom, J. (2000). Andrews’ diseases of the skin. Philadelphia, PA: Elsevier.
Bolognia, J. L., Jorizzo, J. L., Rapini, R. P., et al. (2008). Dermatology. Spain: Mosby.
Habif, T. P. (2004). Clinical dermatology. China: Mosby.
Lever, W. F. (2009). Histopathology of the skin. China: Lippincott Williams & Wilkins.