CORRECT DIAGNOSIS:
Pustular Cutaneous T-Cell Lymphoma
DISCUSSION:
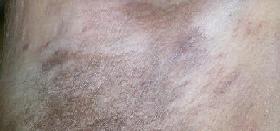
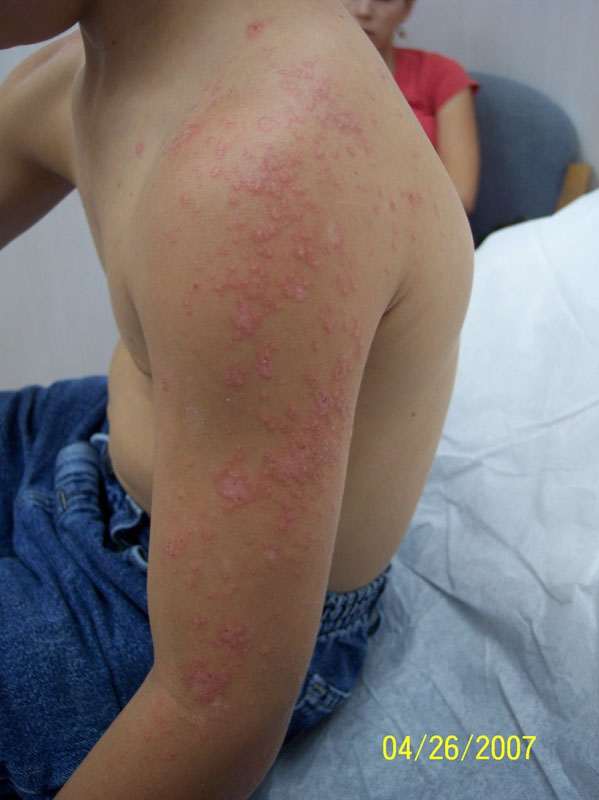
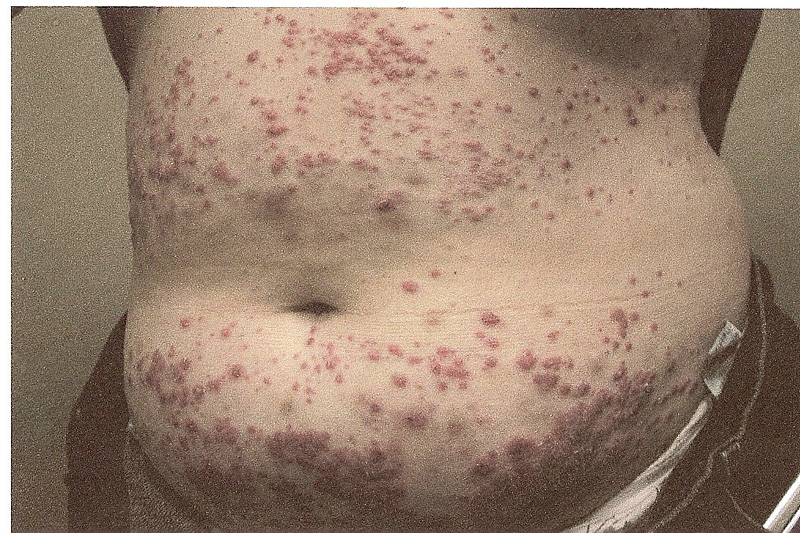
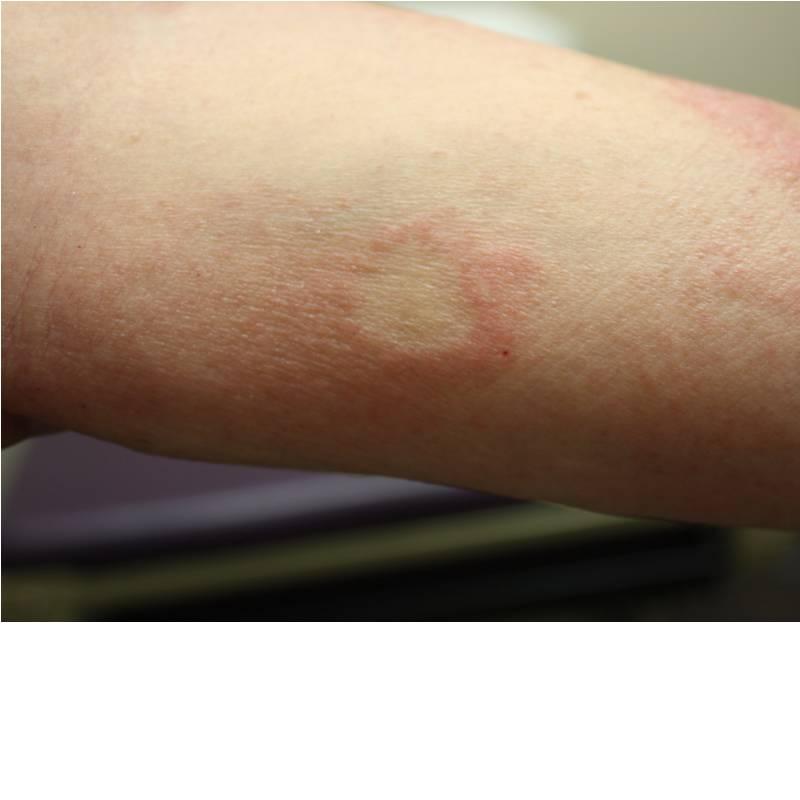
Aside from typical MF, a number of more atypical variants exist including follicular, syringotropic, bullous/vesicular, granulomatous, hypopigmented, hyperpigmented, poikilodermic, hyperkeratotic, vegetating, ichthyosiform, and pustular MF. The patient in the case described above presented with pustular MF, a variant of CTCL marked by localized or generalized non-distinct pustular eruptions. In our particular instance, the patient morphed from a Sneddon-Wilkinson-like presentation of arciform, serpiginous patches and plaques with minute pustules to more circular plaques with peripheral pustules, resembling a pustular psoriasis variant. The persistent nature of the pustular eruption and its resistance to powerful oral therapies was perhaps the only clinical clue that pointed toward a more ominous diagnosis, such as CTCL.
The diagnosis of pustular CTCL is extremely rare, with very few cases documented to date. Biopsy findings are congruent with classic CTCL demonstrating a band-like infiltrate consisting of atypical lymphocytes with cerebriform nuclei and epidermotropism in addition to sub and intra-corneal pustules. Loss of antigen markers occurs as the disease progresses. In our case, reviewing the initial biopsies of perfect subcorneal pustules did reveal rare examples of dermal mononuclear lymphocytes clouded by a heavy neutrophilic infiltrate. The dense collections of neutrophils were deceiving.
Treatment for pustular CTCL is similar to other variants and depends on the extent of disease. The therapeutic resolution has been reported with topical corticosteroids, PUVA, and dapsone. However, our patient was unresponsive to steroids and high doses of dapsone. For more extensive disease, systemic chemotherapy, photophoresis, or biologic treatment with oral bexarotene, retinoids, and interferon have been suggested.
In summary, this case illustrates a rare variant of CTCL which mimicked Sneddon-Wilkinson Disease both clinically and histologically. The persistent nature of the pustular eruption and its resistance to powerful oral therapies was perhaps the only clinical clue that pointed toward a more ominous diagnosis.
TREATMENT:
The patient was started on UVB therapy three times weekly as well as daily oral bexarotene. Significant improvement was observed with currently less than 5% TBSA involvement.
REFERENCES:
Bolognia, J. L., Jorizzo, J. L., & Rapini, R. P. (2008). Cutaneous T-Cell Lymphoma. In Dermatology (2nd ed., pp. 1867-1886). Elsevier.
Kim, Y. H., & Hoppe, R. T. (1999). Mycosis fungoides and Sezary’s syndrome. Seminars in Oncology, 16, 276-289. PMID: 10473714
Van Doorn, R., van Haselen, C. W., van Voorst, P. C., et al. (2000). Mycosis fungoides: Disease evolution and prognosis of 309 Dutch patients. Archives of Dermatology, 136, 504-510. https://doi.org/10.1001/archderm.136.4.504 PMID: 10768639
Kazakov, D. V., Burg, G., & Kempf, W. (2004). Clinicopathological spectrum of mycosis fungoides. Journal of the European Academy of Dermatology and Venereology, 18, 397-415. https://doi.org/10.1111/j.1468-3083.2004.00837.x PMID: 15236664
Pabsch, H., Kunze, J., & Schaller, J. (2009). Mycosis fungoides presenting as a pustular eruption. Journal of the American Academy of Dermatology, 61(5), 908-909. https://doi.org/10.1016/j.jaad.2009.05.019 PMID: 19854105
Vergier, B., de Muret, A., Beylot-Barry, M., et al. (2000). Transformation of mycosis fungoides: Clinicopathological and prognostic features of 45 cases. Blood, 95, 2212-2218. PMID: 10698913