CORRECT DIAGNOSIS:
Proteus syndrome
DISCUSSION:
Proteus syndrome (PS) is exceedingly rare with less than 100 cases having been reported in the literature. It is a progressive hamartomatous disorder that manifests as asymmetric, disproportionate overgrowth affecting tissues derived from any germ layer. It was first described in 1979 by Cohen and Hayden. The name “Proteus Syndrome” was designated by Wiedemann et al. in 1983 after the Greek god Proteus. Like Proteus, this syndrome is capable of assuming many forms, which commonly results in misdiagnosis.
Proteus syndrome results from mosaic expression of a post-zygotic somatic mutation in the AKT1 gene. The AKT1 gene helps regulate cell growth and proliferation, survival, glucose metabolism, genome stability, and neo-vascularization. Constitutive activation of the protein underlies the overgrowth and tumor susceptibility in these patients. Mosaicism results in the random distribution of affected tissues and large degree of variation in severity between patients. An early post-zygotic mutation would result in more disease manifestations than a late mutation, because the early somatic cell carrying a mutation would give rise to more affected cell lineages.
Some reports have suggested that mutations in the PTEN gene are associated with Proteus syndrome. However, it is now believed that individuals with PTEN gene mutations and asymmetric overgrowth do not meet the diagnostic criteria for Proteus syndrome. Instead, these individuals actually have a condition that is considered part of a larger group of disorders called PTEN hamartoma tumor syndrome. One name that has been proposed for the condition is segmental overgrowth, lipomatosis, arteriovenous malformations, and epidermal nevus (SOLAMEN) syndrome; another is type 2 segmental Cowden syndrome. Some articles still refer to PTEN-related Proteus syndrome. AKT1 is activated by loss-of-function mutations in PTEN, which explains why patients with such mutations (those with the SOLAMEN syndrome) and patients with activating mutations in AKT1 (those with the Proteus syndrome) have overlapping clinical manifestations.
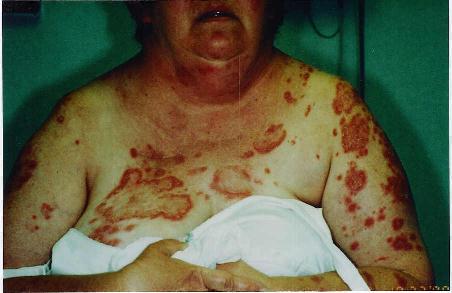
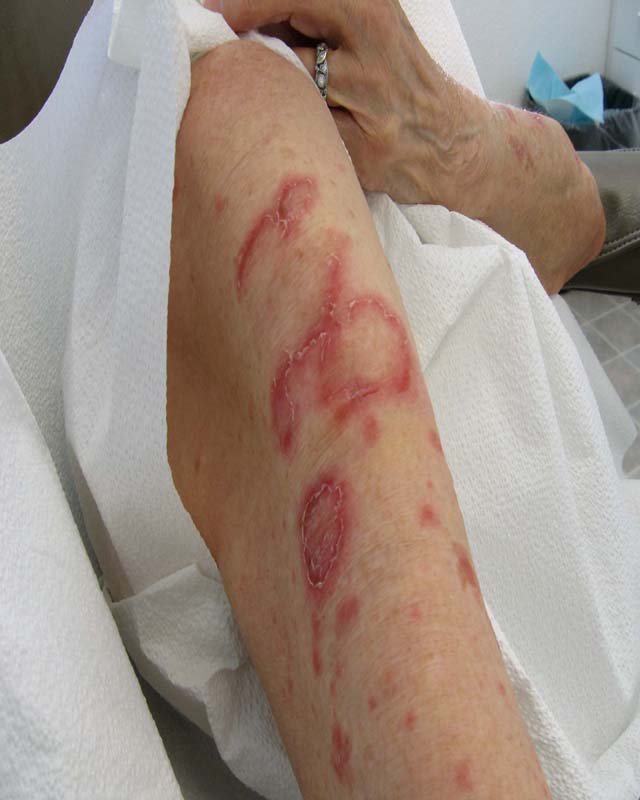
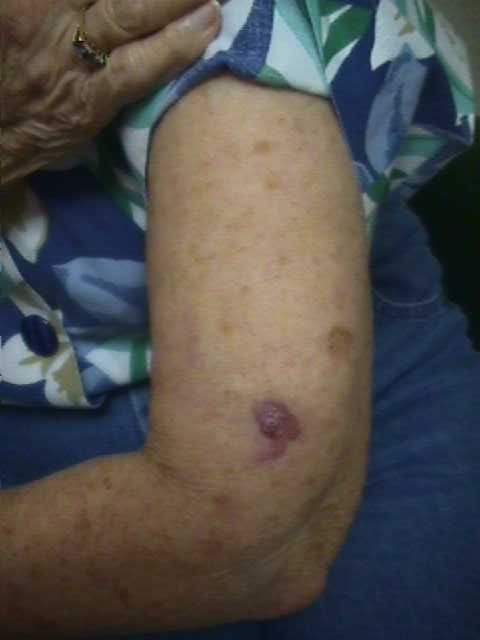
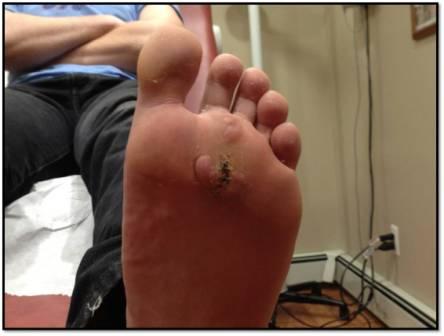
The disease is characterized by postnatal irregular, asymmetric, distorting and progressive overgrowth that can involve many tissues, but most commonly affects bone, connective tissue and fat. Gigantism of the hands and/or feet and hemihypertrophy (partial or complete)is often seen. The cerebriform connective tissue nevus (CCTN) is considered almost pathognomonic for PS. This typically presents in the first or second year of life and evolves slowly, in some patients continuing to develop throughout adolescence. They appear as gyriform gross thickenings of cutaneous and subcutaneous tissues, most commonly present on the soles, occasionally on the hands, abdomen, and nose. Dysregulation of fatty tissue is another prominent feature of PS, with both localized overgrowth (“lipomas”) and lipoatrophy in other areas. Linear verrucous epidermal nevi may also be seen and tend to develop in the first year of life.
Other findings include vascular malformations, most commonly cutaneous capillary malformations, but some patients have venous malformations. Patients with PS are also prone to development of certain tumors, most commonly monomorphic adenomas of the parotid gland, ovarian cystadenomas, and various types of testicular tumors. The characteristic facial phenotype is present in a minority of patients, and is more commonly seen in those with cognitive deficits. Features include downslanting palpebral fissures, flattening of the malar bones, relative lengthening of the face, low nasal bridge with wide nostrils, and a persistently open mouth. Other clinical findings include cystic lung disease(9%), scoliosis as a result of local overgrowth creating asymmetrical forces on the spine(60%), ophthalmologic findings such as strabismus, epibulbar cysts, and epibulbar dermoids(42%), and central nervous system abnormalities, most commonly seizures(40%) and mental deficiency(30%).
The diagnosis of Proteus syndrome is based on clinical findings. Individuals must meet all of the general criteria including mosaic distribution of lesions, sporadic occurrence, and progressive course, along with specific criteria from Categories A-C (either A, two from B, or three from C). Category A contains the cerebriform connective tissue nevus. Category B includes linear epidermal nevus, asymmetric, disproportionate overgrowth, and specific tumors before the second decade (bilateral ovarian cystadenoma, parotid monomorphic adenoma). Category C incudes dysregulated adipose tissue, vascular malformations, the characteristic facial phenotype.
Although PS is primarily a clinical diagnosis, molecular genetic testing for the somatic mutation in AKT1 gene can be helpful to confirm the diagnosis. This can be technically challenging because blood is not an appropriate source and tissue may show low-level mosaicism. Skin scrapings from epidermal nevi in PS patients have been shown to be a good source of mutant cells, and provide an alternate source for genetic testing. It is important to note that PS is not inherited, so prenatal testing is not indicated. Among the differential diagnosis for Proteus Syndrome are those entities described as part of PTEN hamartoma tumor syndrome. Bannayan-Riley-Ruvalcaba syndrome is an autosomal dominant disorder characterized by macrocephaly, angiomatosis, lipomatosis, polyposis of the colon and rectum, and pigmented macules of the penis. These patients lack the progressive digital overgrowth, skull exostoses, epidermal nevi, and palmar or plantar changes seen in the Proteus syndrome. Patients with Cowden syndrome typically present with facial trichilemmomas, acral keratoses, papillomatous lesions, lipomas, hemangiomas, and epidermal nevi (Cowden nevus), but do not develop cerebriform connective tissue nevi. These patients also carry an increased risk for breast thyroid, and endometrial cancer. Proteus-like Syndrome refers to individuals with significant clinical features of PS, but do not meet the diagnostic criteria for PS. They are distinguished by macrocephaly, marked lipohypertrophy, and lack of progressive bony overgrowth.
In segmental overgrowth, lipomatosis, AVMs, epidermal nevus (SOLAMEN) syndrome, patients display thickening of the soles and increased wrinkling instead of the gyri found in CCTN. There is segmental proportionate overgrowth with soft tissue hypertrophy and ballooning effect, as well as lymphatic and shunting arteriovenous malformations. Proteus syndrome may be distinguished from neurofibromatosis by the absence of multiple cafe au lait macules, Lisch nodules, axillary freckling, and multiple neurofibromas. Hemihyperplasia and multiple lipomatosis syndrome (HHML) is characterized by subcutaneous lipomatosis and asymmetric overgrowth (hemihyperplasia) which is not as progressive as PS.
Syndromes characterized by vascular malformations may also be considered in the differential diagnosis of PS. In Maffucci’s syndrome enchondromatosis, most commonly of the hands and feet, with multiple cavernous hemangiomas are seen. This should not be difficult to distinguish from Proteus syndrome owing to the lack of enchondromatosis in Proteus syndrome. Klippel-Trenaunay syndrome is characterized by the three main features of nevus flammeus (port-wine stain), venous and lymphatic malformations, and soft-tissue hypertrophy of the affected limb. There are no CCTN seen and overgrowth is present at birth and more severe than PS. In Parkes Weber, a mutation in the RASA1 gene leads to multiple capillary malformations, including AV fistulas that can lead to heart failure, as well as overgrowth of one limb, most commonly the leg.
In the differential diagnosis of the CCTN is isolated plantar collagenoma, a hamartomatous lesion consisting of proliferation of normal collagen tissue. Collagenomas are commonly encountered in other genetic disorders, such as Busche-Ollendorff syndrome, a rare autosomal-dominant condition, resulting from nonsense mutation in the LEMD3 gene which encodes for a potent negative regulator of bone morphogenic protein and transforming growth factor-¦Â signaling pathways. Recently, a mutation in LEMD3 has been reported also in familial cutaneous collagenomas. Histopathologically, cerebriform connective tissue nevi are characterized by an irregular proliferation of highly collagenized fibrous tissue. Biopsies of lipomatous overgrowths reveal nonencapsulated lobules and mature adipocytes. Vascular malformations are lined by flat endothelium, exhibiting a normal, slow rate of turnover. The flat, organoid type of epidermal nevus in PS shows acanthosis, hyperkeratosis, and papillomatosis.
The sporadic and unpredictable nature of Proteus Syndrome can pose a challenge for health care providers. Since PS can affect many different parts of the body in varying degrees, a multidisciplinary approach is important in the management and prevention of secondary complications. Serial clinical photography with an initial skeletal survey and targeted follow up radiographs should be performed to evaluate the degree of obstruction or deformation based on the patient’s medical history and physical exam. Other imaging recommendations include intracranial MRI to evaluate for CNS malformations that may be associated with developmental delay, mental retardation, or seizures. Findings may include multiple meningiomas, polymicrogyria, and periventricular heterotopias. Abdominal MRI, even in the absence of symptoms is recommended to exclude intra-abdominal lipomas. If present, these can behave aggressively, invading adjacent structures. CT of the chest to evaluate pulmonary cystic malformations should be carried if clinically warranted.
One of the most common causes of death for patients with PS is deep venous thrombosis and pulmonary embolism, even in young children. For this reason, perioperative anticoagulant prophylaxis recommended. This may prove difficult to implement because patients with PS may have vascular anomalies that result in postoperative bleeding. Chronic anticoagulation is not recommended for PS patients since there are no data to assess the risks and benefits, and long-term risks may be high in the setting of vascular anomalies. Tumor surveillance is not recommended in PS patients. While the association with several tumor types is strongly suspected, it is not proven, and the predisposition appears to be broad. There is, therefore, no practical and effective method for screening, and there has been no data to demonstrate that early detection of tumors in PS improves prognosis.
Cerebriform connective tissue nevus (CCTN) is a common dermatologic overgrowth that is usually found at the plantar aspect of the foot. The grooves in CCTN can be difficult to clean leading to the accumulation of bacteria and fungus that may cause infection and a malodor. CCTN can progressively increase in size, grow on previously non-involved areas of the foot and then coalesce. This can be disfiguring, painful, and cause walking impairments. Surgical removal of CCTN can lead to disappointing results since recurrence and painful scarring is possible. Dermatological follow-ups and the use of custom orthotics to manage pain, pressure ulcerations, and/or skin breakdown are preferred treatments. In addition, patients and their families can undergo a great deal of stress from this disease. Clinicians are recommended to assess psychosocial issues routinely with parents and children, encourage counseling if needed, and refer the families to peer support groups.
Regarding progression of skin lesions, Beachkofsky, et al. evaluated 36 patients with Proteus syndrome with serial photography for an average of 53 months. Cerebriform connective tissue nevi showed progression in 13 children but not in 3 adults. Lesions progressed by expansion into previously uninvolved skin, increased thickness, and development of new lesions. Lipomas increased in size and/or number in 8/10 children. Epidermal nevi and vascular malformations generally did not spread or increase in number. Long-term prognosis varies across patients. Approximately 20% of PS patients suffer premature death, most commonly due to venous thromboembolism or pulmonary embolism, pneumonia, or surgical complications.
In conclusion, Proteus Syndrome is a complex disease that can involve many areas of the body, especially the skeletal system, connective tissue, fat, and central nervous system. The variable clinical presentation, rarity of the disorder, and clinical overlap with several other diseases has led to significant confusion and misdiagnosis. Molecular genetic testing can be performed, with the highest yield from epidermal nevi or tissue specimens. Patients should be managed with a multidisciplinary approach.
TREATMENT:
The patient was referred for genetic testing for AKT1 and PTEN gene mutations. Serial debulking procedures have been performed to help with the patients difficulty with ambulation. This has not provided any substantial relief, however, due to continued growth of the lesion and development of new lesions.
REFERENCES:
Cohen Jr, M. M., & Hayden, P. W. (1979). A newly recognized hamartomatous syndrome. Birth Defects Orig Artic Ser, 15, 291-296. PMID: 479799
Wiedemann, H. R., Burgio, G. R., Aldenhoff, P., Kunze, J., Kaufmann, H. J., & Schirg, E. (1983). The proteus syndrome. Partial gigantism of the hands and/or feet, nevi, hemihypertrophy, subcutaneous tumors, macrocephaly or other skull anomalies and possible accelerated growth and visceral affections. Eur J Pediatr, 140, 5-12. PMID: 6881416
Biesecker, L. G., Happle, R., Mulliken, J. B., Weksberg, R., Graham, J. M. Jr, Viljoen, D. L., & Cohen, M. M. Jr. (1999). Proteus syndrome: Diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet, 84, 389-395. PMID: 10028712
Turner, J. T., Cohen, M. M. Jr, & Biesecker, L. G. (2004). Reassessment of the Proteus syndrome literature: application of diagnostic criteria to published cases. Am J Med Genet A, 130A(2), 111-122. PMID: 15308295
Lindhurst, M. J., et al. (2011). A mosaic activating mutation in AKT1 associated with the proteus syndrome. N Engl J Med, 365, 611-619. PMID: 21732820
Altomare, D. A., & Testa, J. R. (2005). Perturbations of the AKT signaling pathway in human cancer. Oncogene, 24, 7455-7464. PMID: 16288233
Blumenthal, G. (2008). PTEN hamartoma tumor syndromes. Eur J Hum Genet, 16, 1289-1300. PMID: 18802493
Cohen, M. M. (2005). Proteus Syndrome: An Update. Am J Med Genet C Semin Med Genet, 137C, 38-52. PMID: 16299719
Dietrich, J., et al. (1998). The Proteus Syndrome: CNS Manifestations. Am J Neuroradiol, 19, 987-990. PMID: 9596206
Juntilla, M. M., et al. (2010). AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood, 115(20), 4030-4038. PMID: 20160064
Wieland, I., Tinschert, S., & Zenker, M. (2013). High-level somatic mosaicism of AKT1 c.49G>A mutation in skin scrapings from epidermal nevi enables non-invasive molecular diagnosis in patients with Proteus syndrome. Am J Med Genet A, 161A, 889-891. PMID: 23553324
Caux, F. (2007). Segmental overgrowth, lipomatosis, arteriovenous malformation and epidermal nevus (SOLAMEN) syndrome is related to mosaic PTEN nullizygosity. Eur J Hum Genet, 15, 767-773. PMID: 17334289
Nelson, A. (2008). Isolated plantar collagenoma not associated with Proteus syndrome. J Am Acad Dermatol, 58, 497-499. PMID: 18261981
McCuaig, C. (2012). Connective tissue nevi in children: institutional experience and review. J Am Acad Dermatol, 67, 890-897. PMID: 22939933
Cohen, M. M. Jr. (1995). Putting a foot in one’s mouth or putting a foot down: nonspecificity v. specificity of the connective tissue nevus in Proteus syndrome. Proc Greenwood Cent, 14, 11-13.
Nguyen, D., Turner, J. T., Olsen, C., Biesecker, L. G., & Darling, T. N. (2004). Cutaneous manifestations of Proteus syndrome. Arch Dermatol, 140, 947-953. PMID: 15315951
Nazzaro, V., Cambiaghi, S., Montagnani, A., Brusasco, A., Cerri, A., & Caputo, R. (1991). Proteus syndrome: ultrastructural study of linear verrucous and depigmented nevi. J Am Acad Dermatol, 25, 377-383. PMID: 1892765
Biesecker, L. G. (2006). The challenges of Proteus syndrome: diagnosis and management. Eur J Hum Genet, 14, 1151-1157. PMID: 16845161
Guidera, K. J., Brinker, M. R., Kousseff, B. G., Helal, A. A., Pugh, L. I., Ganey, T. M., et al. (1993). Overgrowth management in Klippel-Trenaunay-Weber and Proteus syndromes. J Pediatr Orthop, 13, 459-466. PMID: 8351582
Turner, J., Biesecker, B., Leib, J., Biesecker, L., & Peters, K. F. (2007). Parenting children with Proteus syndrome: Experiences with and adaptation to courtesy stigma. Am J Med Genet A, 143A, 2089-2097. PMID: 17664071
Beachkofsky, T. M., Sapp, J. C., Biesecker, L. G., & Darling, T. N. (2010). Progressive overgrowth of the cerebriform connective tissue nevus in patients with Proteus syndrome. J Am Acad Dermatol, 63, 799-804. PMID: 20538090
Slavotinek, A. M., Vacha, S. J., Peters, K. F., & Biesecker, L. G. (2000). Sudden death caused by pulmonary thromboembolism in Proteus syndrome. Clin Genet, 58, 386-389. PMID: 11094838