CORRECT DIAGNOSIS:
Piebaldism
DISCUSSION:
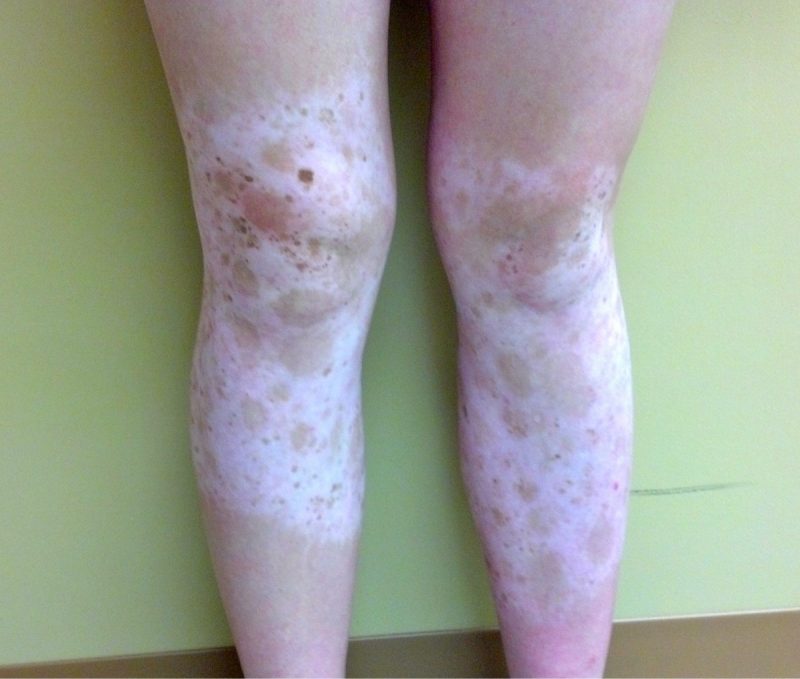
Piebaldism is a rare genodermatosis of autosomal dominant inheritance characterized by a congenital white forelock and leukoderma on the frontal scalp, forehead, ventral trunk, and extremities. Mutations of the c-KIT protooncogene, located on chromosome 4q12, have been associated with the majority of cases of piebaldism. The severity of the phenotype correlates with the type and site of c-KIT mutation.(2)
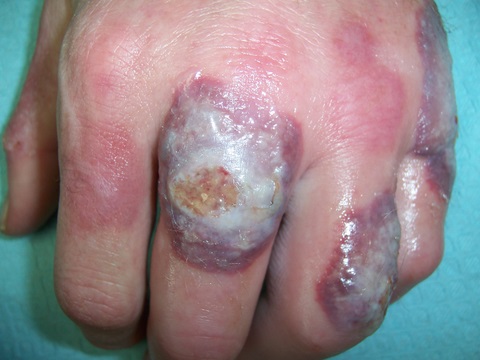
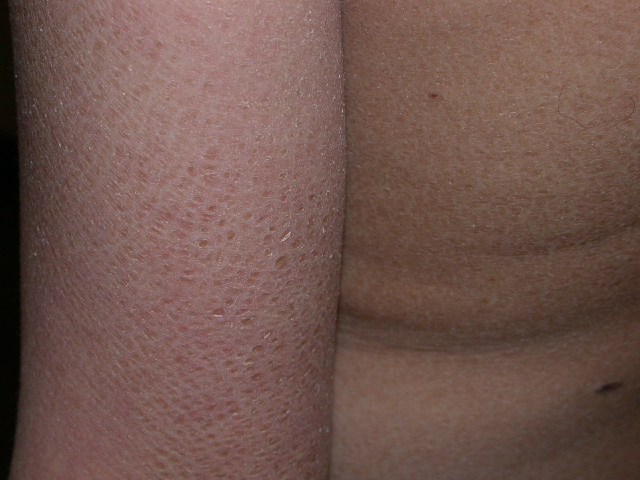
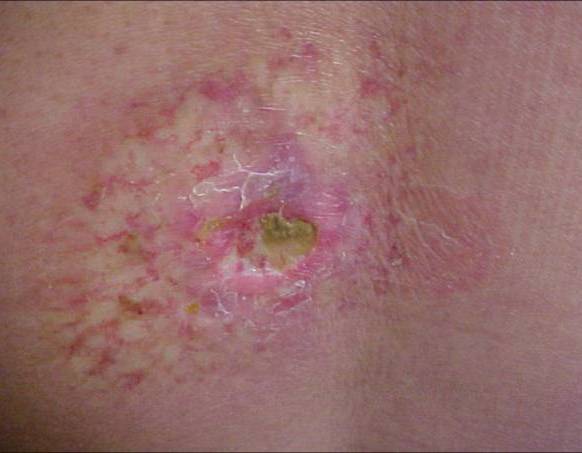
The white patches of piebaldism are present at birth and are relatively stable throughout life. Classically, depigmented areas are symmetrically distributed and have islands of hyperpigmentation within and at the border of the depigmented areas. White patches are devoid of melanocytes, while hyperpigmented areas have a normal number of melanocytes. (1)
The c-KIT gene affects the differentiation and migration of melanoblasts, which originate in the dorsal neural crest tube and migrate ventrally.(1,2) The phenotype of piebaldism is dependent on the specific type and location of the mutation in the KIT gene.(2,3)
Differential diagnoses for piebaldism include vitiligo, albinism, and Waardenburg’s syndrome. In contrast to piebaldism, vitiligo is progressive, rarely congenital, and lacks hyperpigmented macules. Albinism is characterized by generalized depigmentation with a normal number of melanocytes. In Waardenburg’s syndrome, mid-facial lesions, heterochromia irides, and deafness are often present.(1,4) Neurofibromatosis Type I has rarely been associated with piebaldism.(5-6)
Although piebaldism is a benign disorder, it may have a significant psychological impact.(4) Treatment typically includes camouflaging and grafting.(2) A combination of dermabrasion and grafting of pigmented skin into depigmented areas has shown some benefit in selected patients. Unfortunately, there is little benefit from phototherapy alone.(1) Further progress in treatment options is needed to improve the quality of life for individuals with this condition.
TREATMENT:
The clinical diagnosis of piebaldism was discussed at length with the patient. Due to the benign nature of her condition, and patient preference, no further treatment was pursued. She was recommended to use sunscreen daily for the protection of the depigmented areas.
REFERENCES:
Agarwal, S., & Ojha, A. (2012). Piebaldism: A brief report and review of the literature. Indian Dermatology Online Journal, 3(2), 144-147. https://doi.org/10.4103/2229-5178.99802
Oiso, N., Fukai, K., Kawada, A., et al. (2013). Piebaldism. Journal of Dermatology, 40(5), 330-335. https://doi.org/10.1111/1346-8138.12106
Ward, K. A., Moss, C., & Sanders, D. S. (1995). Human piebaldism: Relationship between phenotype and site of kit gene mutation. British Journal of Dermatology, 132, 929-935. https://doi.org/10.1111/j.1365-2133.1995.tb17773.x
Janjua, S., Khachemoune, A., & Guldbakke, K. (2007). Piebaldism: A case report and a concise review of the literature. Cutis, 80(5), 411-414.
Angelo, C., Cianchini, G., Grosso, M. G., Zambruno, G., Cavalieri, R., & Paradisi, M. (2001). Association of piebaldism and neurofibromatosis type I in a girl. Pediatric Dermatology, 18, 490-493. https://doi.org/10.1046/j.1525-1470.2001.18110.x
Duarte, A. F., Mota, A., Baudrier, T., Morais, P., Santos, A., & Cerqueira, R., et al. (2010). Piebaldism and neurofibromatosis type I: Family report. Dermatology Online Journal, 16, 11.