CORRECT DIAGNOSIS:
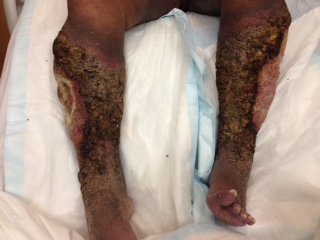
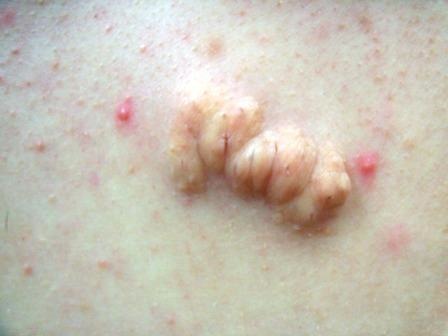
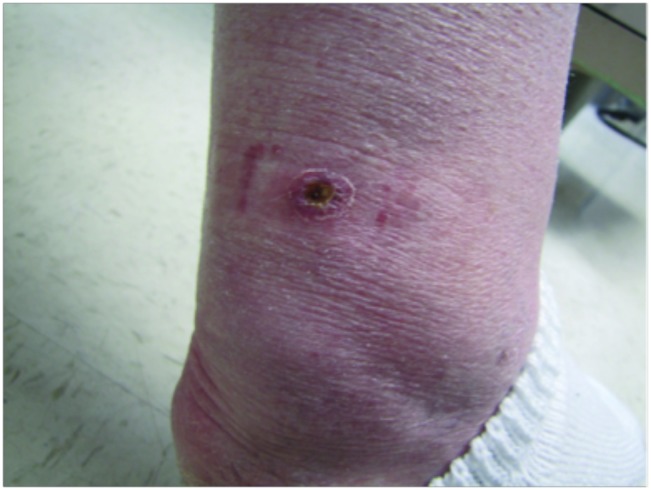
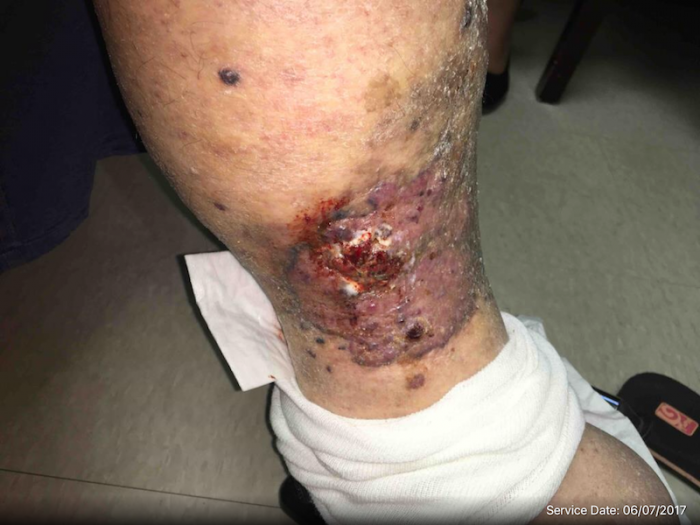
Angiosarcoma
DISCUSSION:
The unfortunate diagnosis of angiosarcoma in the setting of long-standing, significant lymphedema fits the criteria for diagnosis of Stewart-Treves syndrome. Although by definition, Stewart-Treves syndrome involves any malignancy arising in the setting of a chronic lymphedema, the classic case arises in the setting of prior radical mastectomy. The condition itself is rare, with an estimated 400 reported cases worldwide; from a broader perspective sarcomas only represent 1% of malignant tumors and current literature suggests the classic case of Stewart-Treves arising status post radical mastectomy with accompanying lymph node dissection only occurs in 0.07-0.5% of patients. Upwards of 90% of Stewart-Treves syndrome cases are reported to arise in the post-mastectomy patient. Our patient presents not only with the uncommon condition, but also with the less frequently described variety involving a lower extremity in association with lymphedema secondary to radical total abdominal hysterectomy and bilateral salpingo-oophorectomy over 50 years prior.
It is thought that the angiosarcoma arises from the edematous component of the condition rather than the existence of the prior malignancy itself. Literature suggests that the chronic lymphatic congestion results in diminished local immunity, cytokine release, excess cellular degeneration, release of vascular endothelial growth factors (VEGF), and the subsequent development of lymphangiomatosis.
The condition is most commonly reported in the upper extremities of the female population, with peak incidence typically between the ages of 50 – 70 years. In one study, which reviewed 62 cases of angiosarcoma arising in the setting of lymphedema, the average age of onset was 68.8 years. Approximate time between the original surgical intervention and the development of angiosarcoma in the affected extremity is reported to be 10 years. Our case highlights the uncommon development of an angiosarcoma greater than five decades post-operatively.
Clinically, patients present with rapid development of the malignant lesions. Lesions are commonly described as violaceous, and may include macules, papules, and plaques, in a chronically edematous extremity, with or without ulceration. Satellite lesions and telangiectasia may be appreciated as well.
Histologically, the tissue will demonstrate a vascular, poorly formed tumor with extensive infiltration of the dermis and subcutis. In addition, irregular interconnecting vessels and crowding of atypical hyperchromatic endothelial cells may be seen. Solid nodular portions of tumors often demonstrate atypical epithelioid cells. The lesion is classically CD 31 and CD 34 positive, which was demonstrated in our patient.
The malignancy is known to be highly aggressive, with frequent re-occurrence rates and early metastasis. Treatment options include less commonly wide excision, and more commonly limb amputation, with or without adjuvant radiotherapy. Chemotherapy is used sparingly. Prognosis remains dismal. Median survival time is 19 months, a number which literature suggests is slightly improved by early amputation. Common causes of mortality include metastatic lesions to the lungs and chest wall.
TREATMENT:
The patient’s diagnosis, management, and prognosis were discussed with the her in detail. She was referred to surgical oncology for evaluation and determination of further treatment. Further imaging was performed which showed metastasis to the pelvis, therefore the patient was not a candidate for local surgical intervention. She is now receiving palliative care.
REFERENCES:
Stewart, F. W., & Treves, N. (1948). Lymphangiosarcoma in post-mastectomy lymphedema. Cancer, 1(1), 64-81. https://doi.org/10.1002/1097-0142(194801)1:1<64::AID-CNCR2820010108>3.0.CO;2-M
Sathia Raj, V., Ghosh, S., & Chatterjee, S. (2018). Stewart-Treves syndrome: A rare complication of chronic lymphedema. International Journal of Surgery Case Reports, 42, 36-39. https://doi.org/10.1016/j.ijcsr.2018.09.021
Baker, D. J., Deneve, J. L., & Hwang, C. S. (2020). Angiosarcoma in the setting of lymphedema: A review of the literature. Journal of Surgical Oncology, 121(5), 799-806. https://doi.org/10.1002/jso.25834
Gonzalez, M., Rosas, C. G., & Alcaraz, M. A. (2017). Angiosarcoma of the lower extremities in the setting of chronic lymphedema: A case series. The American Journal of Surgery, 213(3), 507-511. https://doi.org/10.1016/j.amjsurg.2016.07.042
Zhuang, Y., & Tseng, L. M. (2016). Histological features of angiosarcoma. American Journal of Dermatopathology, 38(7), 510-515. https://doi.org/10.1097/DAD.0000000000000574
Mason, M. C., Hwang, C. S., & Cormier, J. N. (2015). The epidemiology and treatment of angiosarcoma. Current Oncology Reports, 17(10), 31. https://doi.org/10.1007/s11912-015-0480-5