CORRECT DIAGNOSIS:
Bullous SLE
DISCUSSION:
Epidermolysis bullosa acquisita (EBA), also known as acquired epidermolysis bullosa and dermolytic pemphigoid, is a rare, acquired, subepidermal bullous disease characterized by autoimmunity to type VII collagen, which is a major component of the anchoring fibrils in the DEJ. Presentation may be similar to dystrophic epidermolysis bullosa, bullous pemphigoid, or mucous membrane pemphigoid. EBA has been reported to occur in both children and adults, but more commonly occurs in adults. As one of the rarest of the subepidermal bullous diseases, it occurs with an incidence 0.25/1,000,000 patients, but may be slightly more common among Korean/Asian patients and African Americans.

The pathogenesis is thought to be immune-mediated with both tissue bound and circulating IgG autoantibodies to Type VII collagen (specifically the NC1 domain). This may result in
interference with assembly of type VII collagen and its integration into anchoring fibrils or its ability to interacts with matrix proteins (laminin 5). EBA is associated with certain class II MHC haplotypes (DRB1-1501, DR5, DRB1-13) which likely have a critical genetic impact on susceptibility to developing EBA.
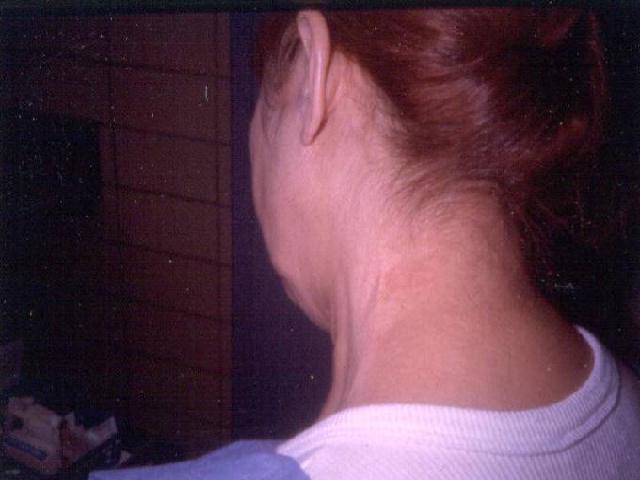
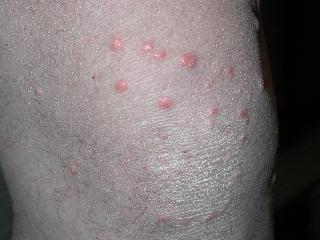
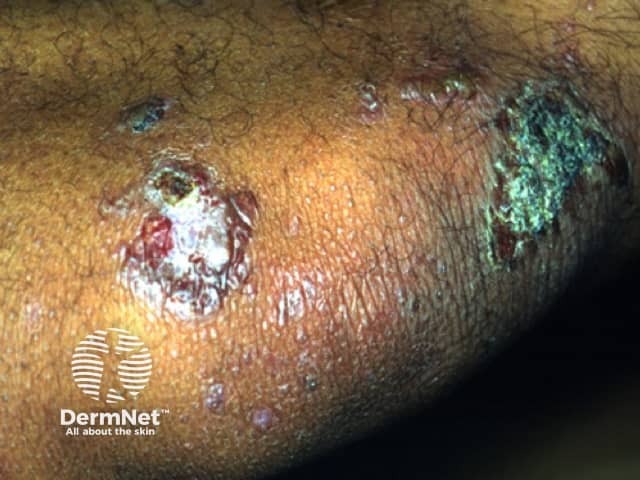
The classical clinical presentation is acral blisters which heal with atrophic scarring, milia, and hyper- or hypo- pigmentation. There tends to be no background inflammation to the skin. The blister fluid is typically serous, but can be hemorrhagic. The areas may eventually progress to erosions. The most prominent areas tend to be found in trauma prone areas (elbows, knees, dorsal aspects of hands, feet, and toes). Unfortunately, the scarring can be mutilating, leading to the development of mitten deformity, syndactyaly, nail dystrophy, or complete nail loss. Other variants of the clinical presentation include bullous pemphigoid-like lesions with bullae involving intertrigenous and flexural areas that heal without milia or atrophic scarring. Mucous membrane pemphigoid-like presentation can also occur with erosions and intact vesicles in the mouth, larynx, and esophagus, which can lead to dysphagia and laryngeal stenosis. During the course of the disease, it can convert from one variant to another or become a mixed presentation.
The onset of EBA may be associated with multiple systemic disease including inflammatory bowel disease, multiple myeloma, systemic lupus erythematosus, rheumatoid arthritis, thyroiditis, and diabetes mellitus. In patient with both EBA and inflammatory bowel disease, Crohn’s disease occurs more frequently than ulcerative colitis.
TREATMENT:
Epidermolysis Bullosa Acqusitica is a rare disease and as a result there has been limited large scale clinical trials. An important aspect in the management of EBA is avoidance of physical trauma. Patients should avoid hot water and harsh cleansers. An appropriate initial treatment is to start the patient on a systemic corticosteroid combined with dapsone or mycophenolate mofetil. In certain adult patients corticosteroids can be given with both dapsone and mycophenolate mofetil. Other drugs that have been tried with some success include colchicine, cyclosporine, IVIG, extracorporeal photophoresis and even rituximab.
REFERENCES:
1. Bernard P, Vaillant L, Labeille B, et al: Incidence and distribution of subepidermal autoimmune bullous skin diseases in three French regions. Bullous Diseases French Study Group. Arch Dermatol 1995; 131: pp. 48-52
2. Chen M, Kim GH, Lori Prakash BS, and Woodley DT: Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity 2012; 45: pp. 91-101
3. Sitaru C: Experimental models of epidermolysis bullosa acquisita. Exp Dermatol 2007; 16: pp. 520-531
4. Kim JH, Kim YH, and Kim SC: Epidermolysis bullosa acquisita a retrospective clinical analysis of 30 cases. Acta Derm Venereol 2011; 91: pp. 307-312
5. Briggaman RA, Gammon WR, and Woodley DT: Epidermolysis bullosa acquisita of the immunopathological type (dermolytic pemphigoid). J Invest Dermatol 1985; 85: pp. 79s-84s
6. Gammon WR, Briggaman RA, Inman AO, et al: Differentiating anti-lamina lucida and anti-sublamina densa anti-basement zone antibodies by indirect immunofluorescence on 1.0M sodium chloride-separated skin. J Invest Dermatol 1984; 82: pp. 139-144
7. Lebwohl, M.G. et al. (2014). Epidermolysis Bullosa Acquisita in Treatment of Skin Disease (pp.213-217).Elsevier
8. Image: https://dermnetnz.org/topics/epidermolysis-bullosa-acquisita